
Síndrome de Prader Willi e Angelman: Causas, Sintomas e Tratamentos
Descubra como diagnosticar e tratar a Síndrome de Prader Willi e Síndrome de Angelman. Obtenha informações sobre os sintomas, tratamentos e pesquisas em andamento para ajudar você a lidar com esses distúrbios genéticos.



Síndrome de Prader Willi
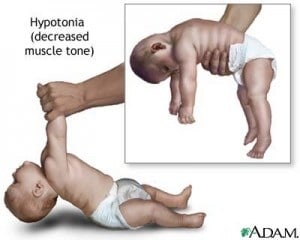
Esta síndrome, descrita em 1956, é de origem genética localizada no cromossomo 15, ocorrendo no momento da concepção. Afeta meninos e meninas em um complexo quadro de sintomas, durante toda a sua vida, estes variando em presença e intensidade de indivíduo para indivíduo. Um diagnóstico precoce, antes da manifestação dos sintomas, principalmente a obesidade, tem trazido uma melhora na qualidade de vida dos portadores nos últimos anos. Mas ainda em caso de diagnósticos tardios, uma série de cuidados pode ser iniciada, retornando na qualidade de vida dos portadores.
Bebês com Síndrome de Prader Willi (PWS) apresentam baixo Apgar ao nascer, dificuldade de sugar, choro fraco e são muito pouco ativos, dormindo a maior parte do tempo. Raramente conseguem ser amamentados. Seu desenvolvimento neuro-motor é lento, tardam a sentar, engatinhar e caminhar.
Os sintomas da síndrome variam de indivíduo para indivíduo e estão também associados ao ambiente em que este vive, aos estímulos e ao acompanhamento terapêutico e educacional que recebe. Os principais sintomas são:
|  |
A obesidade manifestada por muitos portadores é consequência de um consumo excessivo de calorias, pelo comportamento compulsivo em relação à comida, somado a fatores metabólicos e pouca atividade física. A fome constante é provavelmente causada por uma desordem do hipotálamo, no cérebro: durante uma refeição, a “mensagem” de saciedade não é processada. E se não controlado esse acesso à quantidade/composição da comida, o ganho de peso é rápido. Portadores desta síndrome, em geral, necessitam de algum nível de assistência ou supervisão em sua alimentação, mesmo quando bem informados de sua condição de saúde. Diferentemente, por exemplo, de portadores de diabetes.
Muitos efeitos relacionados aos sintomas podem ser amenizados com um diagnóstico, que proporciona a chance de intervenções terapêuticas e educacionais; pelo conhecimento e compreensão da síndrome pela família, que deve buscar estruturar um ambiente inclusivo, seguro, assistido e estimulador para o indivíduo se desenvolver; e por um acompanhamento de saúde e educação adequada.

Bibliografia:
- PWS ASSOCIATION UK – Babies and children with Prader-Willi Syndrome 2001 – ISBN- 0 9521023 6 6
- BALLONE, G.J. – Transtornos Alimentares, in. PsiqWeb, no endereço eletrônico <http://www.psiqweb.med.br/anorexia.html>, revisto em 2003.
- ASOSIACION ESPANOLA PRADER-WILLI, Guía familiar y profesional del síndrome de PRADER-WILLI. Cada capítulo está escrito por um especialista no tópico. 400 p.; 2001.Fonte: http://www.fiocruz.br
Tratamentos e Cuidados
O tratamento para ambas as síndromes envolve uma abordagem multidisciplinar, incluindo acompanhamento médico, nutricional e terapias específicas. Para saber mais sobre a importância da educação inclusiva e como ela pode beneficiar crianças com necessidades especiais, consulte nosso artigo.
Além disso, o suporte familiar e a criação de um ambiente seguro e estimulante são fundamentais para o desenvolvimento das crianças afetadas. Para mais informações sobre como lidar com desafios de aprendizagem, você pode conferir nosso texto sobre desafios de aprendizagem.
Por fim, é essencial que os cuidadores estejam atentos às necessidades emocionais e sociais das crianças, promovendo interações que favoreçam o desenvolvimento de habilidades sociais e emocionais. Para dicas sobre como lidar com alunos autistas, acesse nosso artigo sobre como lidar com alunos autistas.
