
Síndrome de angelman
| CARACTERÍSTICAS DA SÍNDROME DE ANGELMAN | |
| Sinal | % |
| Hipotonia | 90% |
| Atraso neuropsicomotor | 100% |
| Deficiência mental grave | 100% |
| Ausência de fala | 98% |
| Convulsões | 80% |
| Ataxia (alterações da marcha)* | 100% |
| Acessos de riso | 90% |
| Microcefalia | 90% |
| Macrostomia (boca grande) | 75% |
| Prognatismo | 95% |
| Dentes espaçados | 60% |
| Sialorréia (baba) | 70% |
| Protrusão de língua | 70% |
| * A maioria dos afetados começa a caminhar entre 3 e 4 anos de idade | |
HISTÓRICO
Foi o Dr. Harry Angelman quem descreveu o quadro clínico pela primeira vêz em 1965. Os sintomas comuns das três criancas eram: andar desajeitado, risadas frequentes, convulsões e perímetro cefálico pequeno (microcefalia) e achatamento occiptal (braquicefalia).
O autor, inicialmente denominou esta Síndrome de “Happy Puppet” (boneca alegre) nome que hoje está em desuso por ser inadequado para descrevr esta síndrome. Este nome foi consequencia da presença de risos e mesmo gargalhadas que são características dessas crianças.
Mais que vinte anos separam a descrição clínica da descoberta de uma relação com a etiologia genética, que se deu em 1988 (1). Nêste trabalho, o autor descreveu a associaço, em alguns pacientes, do quadro de Síndrome de Angelman (SA), com uma deleção intersticial (perda de pequena parte) do braço longo “q”, na região entre 11 e 13 do cromossoma de número 15. Com estudos citogenéticos mais modernos se detecta esta deleção na maioria dos pacientes afetados por esta Síndrome. Hoje admite-se que a ausência desta região do cromossomo 15 proveniente da mãe, é a responsável pela presença do quadro clínico.
INTRODUÇÃO
A SA é de origem genética e, embora clínicamente bem delimitada, é pouco divulgada em círculos profissionais e científicos não genéticos. Tal fato acarreta em desconhecimento por parte dos profissionais da Saúde Mental Infantil (SMI), mesmo os de formação médica, psiquiatras e neurolgistas da infância.
Os afetados apresentam, com muita frequência, sintomas comuns ao grupo nosológico dos Transtornos Invasivos do Desenvolvimento (2). Retardo Mental grave, sintomas autísticos, grave comprometimento da fala e mesmo sua ausência, epilepsia e retardo neuro-psico-motor são alguns dos sintomas encontrados na maioria das crianças com SA.
No que se refere a citogenética é sabido que as pessoas normais possuem 23 pares de cromossomas, sendo um dêles o sexual – XX na mulher e XY no homem. Na formação do zigoto humano normal, cada progenitor participa com uma unidade de cada par.
Os cromossomas foram divididos em grupos de acordo com suas características,tamanho e posição do centrômero. Todos possuem dois braços, denominados “p” – curto, e “q” – longo.
No processo evolutivo das técnicas da citogenética,os cromossomas foram bandeados, o que facilitou seu reconhecimento pelos técnicos.
Vejamos a foto do cromosssoma de número 15 em pró-metáfase. O braço “p” apresenta três bandas, numeradas respectivamente de 11 (que possui duas sub-bandas 11.1 e 11.2), 12 e 13.
Vemos, de cima para baixo, no braço “q” a região número 1 com bandas, 11.1 de cor escura,a 11.2 clara, a 12 escura e assim por diante, até a banda clara 15. Seguindo,temos a região 2 com as bandas numeradas de 21.1 a 26.3.
A anomalia responsável pela SA ocorre no espaço entre 11 e 13 do braço “q”. DNA,
Há alguns mecanismos conhecidos que resultam no fenótipo da Síndrome de Angelman.
Um é quando há deleção do cromossoma 15 no braço longo “q” na região entre 11 e 13, que ocorre em aproximadamente 60% (3) dos casos. Se examinarmos êste material verificaremos que a deleção ocorre no cromossoma originário da mãe.
O outro é quando a pessoa recebe os dois cromossomos 15 do Pai, ao que denominamos dissomia, sem porém haver deleção. A frequência dêste mecanismo é abaixo de 10% (4). Nêste caso o diagnóstico só é feito com estudos de DNA.
Outra possibilidade é quando há deleção do DNA (5). Também aqui o dignóstico será através de “estudos de DNA”.
Um mecanismo ocasional, é quando há uma translocação com perda de material desta região, 15q.11-13.
Importante salientar que sòmente no primeiro e no último mecanismo haverá alteração citogenética, ou seja visível em microscópio. Porém, para tanto, é necessário que o exame seja realizado com técnica especial, em pró-metáfase onde o cromossoma “está comprido” (ainda não se condensou). Enquanto os cromossomas, em prometáfase exibem de 550 a 850 bandas, só visualizamos cerca de 400, quando preparado em metáfase. Tal questão é de importância prática para o clínico, pois êle deve solicitar a técnica de “cromossoma longo” ou em pró-metáfase para pesquisa de SA. Caso isto não seja mencionado, assim como a hipótese diagnóstica, certamente esta técnica não será executada o que poderá resultar em falso negativo para SA.
QUADRO CLÍNICO (6,7)
Não há prevalência de sexo entre os afetados. O diagnóstico clínico precoce é raro e difícil. O quadro clínico vai se evidenciando com o passar do tempo, tornando o diagnóstico mais fácil com a evolução da Síndrome.
O fenótipo clássico é caracterizado por:
a) Atraso do crescimento de início pós-natal. Geralmente ao nascimento, o pêso e a estatura estão situados nos percentis normais mas, a estatura final é baixa.
b) A criança, com frequência, apresenta um peculiar aspecto de fragilidade.
c) Existe uma tendência a apresentar a pele, os olhos e os cabelos hipopigmentados na raça branca.
d) Braquicefalia, crânio curto no diâmetro antero-posterior.
e) Microcefalia, geralmente de início pós-natal, embora algumas vêzes possa existir já ao nascimento. Os cabelos tendem a serem esparsos.
f) Pode ocorrer palidez ou atrofia do nervo óptico, estrabismo e presença de manchas de Brushfields na periferia da iris.
g) A boca costuma ser grande e é comum haver a postura de protusão da língua acompanhada de sialorréia. O maxilar tende a ser hipoplásico, enquanto a mandíbula é protusa (prognatismo).
h) Os dentes são usualmente pequenos e espaçados.
i) A presença de episódios de sorrisos e gargalhadas imotivadas são muito comuns nêstes quadros.
j) Há retardo neuro-muscular, geralmente grave. Hipotonia muscular de origem pré-natal, levando a um andar desajeitado e atáxico; hiper-reflexia ocasional; epilepsia na forma de hipsarritmia ou grande mal já nos primeiros anos de vida e o EEG é geralmente anormal, apresentando espículas de alta amplitude.
O fenótipo psicológico ou o quadro clínico psiquiátrico revela:
a) Distúrbios no contato interpessoal de grau leve a moderado e hipercinesia, usualmente grave. Estereotipias de mãos (usualmente como bater palmas seja na linha média do corpo ou não).
b) Cognição a nível sensório-motor (8), comunicação expressiva ausente ou profundamente comprometida – usualmente há comunicação a nível pré-linguístico muito primitiva, ou mais elaborada como apontar para objetos. A linguagem compreensiva tende a ficar mais desenvolvida com a idade.
c) Há bruxismo frequente e auto-agressividade em situações de frustração (morder-se e bater a cabeça).
Frequência das manifestações clínicas na Sindrome de Angelman (9)
- RDPM 100%
- RM grave 100%
- Mutismo ou uso de < 6 palavras 100%
- Ataxia 100%
- Sorrisos frequentes 96%
- Distúrbios de sono 90%
- Epilepsia 86%
- Flapping de mãos 84%
- Dificuldades alimentares 72%
- Estrabismo 42%
- Hipopigmentação em relação a família 39%
- Escoliose 11%
HISTÓRIA NATURAL
Como o recém-nascido não apresenta as características fenotípicas faciais e nem as demais alterações que ficarão evidentes com o tempo, o diagnóstico geralmente só é feito a partir dos primeiros anos.
O período pré-natal, usualmente, é normal assim como o peri-parto. Pêso e estatura geralmente são normais, não sendo comuns ocorrências de malformações ao nascimento.
São frequentes os relatos de dificuldades na alimentação do bebê, sendo que alguns não demonstram interesse, nesta atividade vital.
Também é comum as crianças regurgitarem, fato que leva os familiares a pensar que não estão gostando da alimentação. Por tal motivo são também frequentemente avaliados com suspeita de refluxo gastro-esofágico que, quando presente, raramente é grave.
O sorriso social pode estar presente antes dos 6 meses de idade. Já no período anterior a um ano de idade, pode apresentar as tão características gargalhadas e sorrisos imotivados. A frequente protusão da língua a faz parecer maior que realmente é. O estrabismo é um dos poucos sinais já presentes antes da idade de 1 ano, numa frequência de 30 a 50% dos afetados.
Hipotonia muscular e o atraso motor ficam evidentes na faixa etária próxima a um ano, pois nêstes casos ainda não conseguem sentar sem apoio. Embora algumas crianças nasçam normocefalicas há uma evidente diminuição do crescimento craniano entre 6 a 12 meses (1), verificado a partir da diminuição dos percentis cefálicos.
Entre 1 e 3 anos de idade fica evidente o atraso no desenvolvimento, o que suscitará a necessidade de levar a criança a uma avaliação médica. Já há um quadro de microcefalia e, possívelmente, de epilepsia. Na Tomografia Computadorizada Cerebral pode-se constatar a presença de leve atrofia cerebral. Os exames de “screeening metabólico” são normais assim como as biópsias de nervo e os testes de condução neural. Nêste período apresentam déficit de atenção com intensa hiperatividade e põem práticamente todos os objetos na boca. Na medida que ficam mais excitadas, riem mais e apresentam movimentos de balançar os braços – de uma forma contínua e desajeitada, bater palmas e colocar as mãos na boca.
O caminhar é um dos grandes problemas a serem solucionados, pois, algumas ciancas são tão hipotônicas e atáxicas, que só conseguem a deambulação após muito trabalho. Embora algumas consigam andar com 2 a 3 anos, outras não. Algumas crianças nunca vêm a andar e outras perdem tal aquisição, necessitando de cadeiras de rodas. A escoliose, na adolescência, pode ocorrer nas que não aprenderam a andar.
O desenvolvimento normal da fala não aparece, embora possam surgir fonemas e/ou algumas palavras. A linguagem compreensiva é mais efetiva que a expressiva.
A epilepsia pode surgir antes dos 3 anos de idade e mesmo as crianças que não as tenham, frequentemente apresentam alterações no EEG.
A aquisição das Atividades de Vida Diário (AVD) é de suma importância pois, às vêzes, não há controle esfincteriano mesmo aos 8 anos, sendo que algumas não adquirem nunca o controle esfincteriano noturno. O comer e vestir-se são prejudicados pelos movimentos involuntários dos membros superiores, embora haja uma tendência à sua diminuição na adolescência.
A atividade de dormir é prejudicada pela hiperatividade e deve haver o cuidado especial na utilização de camas de baixa altura ou com proteç¨es, para que a criança não caia. Algumas famílias utilizam meia-porta, como forma de limitar a movimentação delas.
Embora com atraso de 1 a 3 anos, a maturidade sexual ocorre. Mesmo com o passar da idade permanecem com uma fisionomia de adolescentes e as características fenotípicas faciais tendem a se tornar mais marcantes. Alguns afetados desenvolvem a capacidade de falar algumas palavras e alguns poucos apresentam ecolalia retardada, onde podem até cantar trechos de músicas.
Embora não haja ainda trabalhos de significante “follow-up” de afetados com a SA, sabe-se que na adolescência e na idade adulta, nos poucos casos conhecidos, não demonstram ocorrência de maior frequência de patologias como pneumonia e outras infecções quando comparados com a população em geral.
DIAGNÓSTICO
O diagnóstico clínico da SA depende muito da faixa etária em que o paciente é avaliado e, também, da experiência do observador.
O quadro clínico, no primeiro ano de vida, pode ser confundido com outras patologias e só se torna mais evidente entre 1 e 3 anos. Mesmo nesta faixa etária, alguns pacientes não apresentam o fenótipo típico. Pela nossa experiência, o sinal que mais nos parece constante é o distúrbio grave da linguagem, quando associado ao Retardo Mental.
Em nosso meio, o exame complementar mais utilizado é o estudo cromossômico. É feito uma análise dos cromossomos em pró- metáfase (cromossomos longos), com atenção especial ao de número 15. Nele pode ser observado uma deleção intersticial no braço longo – região 11-13, na maioria dos casos da SA.
Esta deleção pode ser confirmada por estudo molecular. A análise do DNA pode também confirmar o diagnóstico, em alguns pacientes que apresentam uma deleção muito pequena, não observada através das técnicas citogenéticas.
Outro exame que pode auxiliar, embora inespecífico, é o EEG (10,11) que pode mostrar as alterações características, mesmo na ausência de episódios comiciais. Também observa-se atrofia cerebral leve através da Tomografia Computadorizada do Crânio e a Ressonância Magnética (12).
DIAGNÓSTICO DIFERENCIAL (DD)
O conhecimento da SA é muito importante para os profissionais da SMI, pelo grande número de patologias que concorrem na lista de DD. A presença precoce do diagnóstico, com certeza, influirá positivamente no prognóstico de nosso pequeno paciente através da instituição do tratamento mais apropriado.
Paralisia Cerebral (PC), Retardo Mental Profundo (RM) de etiologia desconhecida, Síndrome do Autismo Infantil (SAI) e Síndrome de Rett (SR), ocupam o cenário principal do DD da SA.
SA e Paralisia Cerebral
O DD deve-se ao fato da presença de sinais comuns como o Retardo Neuro-Psicomotor – comumente grave, microcefalia, ausência ou fala gravemente comprometida, “aspecto” sugestivo de RM, ataxia, estrabismo, epilepsia e outros sinais neurológicos de dano cerebral. Uma situação que devemos sempre estar atentos, para “levantarmos” a hipótese de SA é, quando o quadro clínico sugere Paralisia Cerebral mas o “APGAR” não foi baixo.
Devemos lembrar que pode ocorrer concomitância de sofrimento fetal e, portanto, clínica de paralisia cerebral, com a SA.
SA e Retardo Mental profundo de etiologia desconhecida
Os portadores de SA diferenciam-se pelo melhor nível de linguagem compreensiva, por apresentarem sinais de dano neurológico e evidentes dismorfismos (alterações do padrão normal do corpo) como microcefalia, braquicefalia, prognatismo, risos sem motivo aparente, baixa estatura, sintomas autísticos e ausência de padrão de RM familiar.
SA e S.Autismo Infantil
É um dos mais interessantes e surpreendentes diagnósticos diferenciais que temos tido a oportunidade de presenciar, tanto pela frequência de diagnósticos que os autores têm realizado como pelas filigranas dos sinais que nos levam a pensar em SA.
O distúrbio no contacto inter-pessoal, a hipercinesia, os graves distúrbios de fala, a defensividade táctil, episódios de auto- agressividade, insatisfação em serem carregados, quando bebês, são sintomas comuns entre estas duas entidades nosológicas. Porém, na SA, o sintoma autismo, quando presente, é comumente de grau leve a moderado.
Não é comum a “mesmice” que vários portadores de SAI possuem, mas pode estar presente. Há o olhar, mas sem fixação no interlocutor. Quando se machuca, chora e busca proteção ou carinho. Apresentam as risadas sem motivo desde pequenos, sendo que, às vêzes, apresentam sòmente esgares de riso. Menor dificuldade de alimentação com sólidos. Desenvolvem muito a capacidade de comunicação compreensiva. Isto tudo sem retomar a presença dos dismorfismos e da “gestalt” específica que os potadores de SA possuem.
Chamamos a atenção para os distúrbios na relação interpessoal (autismo) e o grave distúrbio da fala, comum nas duas entidades. O transtorno de fala é o que nos têm norteado para o significante número de diagnósticos de SA que temos feito. Nossa prática de rastrear citogenéticamente os autistas com ausência de fala, dismórficos ou não, nos têm mostrado maior frequência de deleção dêste segmento do cromossoma 15, que outras patologias, usualmente publicadas como associadas com o Autismo Infantil, como por exemplo a Síndrome do X-frágil.
SA e Síndrome de Rett
Outro DD difícil clínicamente é o de SA e S. Rett (Estágio III entre 2 e 10 anos), tratando-se, exclusivamente de meninas. Em nossa experiência clínica, temos obtido relatos de parada do desenvolvimento, também em portadoras de SA. Em tais casos,embora não sejam mudanças espetaculares, como ocorre na SR clássica, há perda de uso de fonemas, ocorrência de um maior isolamento e hipercinesia. Em ambas patologias, as crianças crescem pouco, embora tenham nascido com tamanho adequado, não falam, apresentam sintomas autísticos, hipercinesia, bruxismo, ataxia, apraxia e epilepsia. A estereotipia de mãos, principalmente a forma de bater palmas muitas vêzes nos dá a impressão de estarmos diante de uma portadora de SR, tão grande é a semelhança. Até a microcefalia pode ser adquirida nos dois quadros.
O “olhar” de busca de comunicação e, as vêzes, de sofrimento que as portadoras de SR possuem, que todos que trabalham com elas conhecem, não se vê nas meninas com SA.
Outra questão que temos presenciado é o prognóstico relativamente favorável que os portadores de SA apresentam em relação as patologias apresentadas nêste rol de diagnósticos diferenciais.
TRATAMENTO
O tratamento da criança com SA inicia-se com um trabalho junto aos Pais, de esclarecimento quanto a aspectos objetivos da Síndrome, assim como aspectos psico-dinâmicos do casal e da família. Frustração, desespero, revolta e aceitação são fases que esperamos que êles vivenciem, para estarem prontos, para permitirem o tratamento do/a filho/a. Todos nós que trabalhamos com crianças, sabemos que, indubitavelmente, o tratamento delas passa pelos Pais. Uma orientação cuidadosa e contínua coloca-os diante de parâmetros objetivos e pode deixa-los mais seguros e com menos sentimento de culpa.
Passado o momento da questão: “O que minha criança têm?” e da devolução, chegamos aos sintomas-alvo mais importantes que dependem da idade da criança. A fisioterapia é necessária para as menores, que ainda não deambulam. Fonoaudiologia é importante para aquelas que têm grave distúrbio da coordenação orofaringeana, que causam dificuldades de deglutição e intensa salivação. A Terapia Ocupacional é importante para todas elas, pois é através desta técnica que estas crianças obterão maior organização corporal e cognitiva para a vida futura.
No que se refere a medicação, o sintoma mais importante a ser trabalhado é o déficit de atenção.
Quando a criança é muito pequena, o arsenal terapêutico médico é restrito. O uso do Cloridrato de Cipro-heptadine acalma, aumenta a atenção voluntária, propicia um sono noturno mais prolongado e melhora o apetite.
O uso de neurolépticos, nos portadores de SA, têm sua indicação restrita pela propensão a impregnação e ao fato que esta medicação, sabidamente diminui a capacidade cognitiva do usuário e o umbral epileptogênico.
As hipercinéticas respondem a Clorpromazina, que deve ser administrada com cuidado. Também a Propericiazina pode ser usada com êste objetivo. Em alguns casos, a Tioridazida traz benefícios importantes.
Quanto aos sintomas autísticos, temos tido bons resultados na administração de Cloridarto de Piridoxina (vitamina B6) em doses altas, sem porém atingir as doses preconizadas para seu uso em SAI (13). Com a Vit.B6 obtêm-se uma franca melhora da atenção voluntária, da utilização da vontade própria, da comunicação compreensiva e, consequentemente, uma melhor performance da memória.

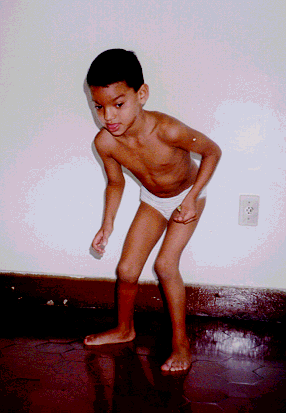

SÍNDROME DE ALGEMAN
O diagnóstico é em geral estabelecido por geneticista ou neurologista, e baseia-se em elementos clínicos, especialmente o retardo do desenvolvimento neuromotor, a ocorrência de crises convulsivas e a presença de características físicas peculiares. Nas crianças que já adquiriram a marcha, chama à atenção o andar bastante desequilibrado, com as pernas abertas e com os membros superiores afastados do corpo, como se tentando melhorar o equilíbrio, e acompanhado de movimentos trêmulos e imprecisos. O comportamento caracteriza-se por expansividade e riso fácil e freqüente. A comunicação é bastante prejudicada em virtude da capacidade reduzida da expressão pela fala. A SA pode ser confundida com deficiência mental de causa indeterminada, autismo infantil ou paralisia cerebral. As características principais, mas não necessariamente presentes, estão relacionadas abaixo:
O que é a Síndrome de Angelman?
A Síndrome de Angelman (SA) é um distúrbio neurológico que causa retardo mental, alterações do comportamento e algumas características físicas distintivas. Ela foi, pela primeira vez, relatada em 1965, quando um neurologista britânico, Dr. Harry Angelman, descreveu 3 crianças com este quadro. Até 1987, o interesse por esta doença foi bastante reduzido. Neste ano, observou-se que a análise dos cromossomos de afetados por SA mostrava, em cerca de 50% dos indivíduos, a falta de uma pequena porção (deleção) do cromossomo 15. O que parecia ser uma situação muito rara mostrou-se bastante freqüente. Estima-se, atualmente, que uma em cada quinze ou vinte mil crianças são afetadas por esta doença.
Como se dignostica a Síndrome de Angelman?
·1 Atraso na aquisição motora (sentar, andar, etc.) ·2 Ausência da fala ·3 Andar desequilibrado, com pernas afastadas e esticadas ·4 Natureza afetiva e risos freqüentes ·5 Sono entrecortado e difícil ·6 Redução do tamanho da cabeça e achatamento de sua porção posterior ·7 Características faciais distintivas: boca grande com protusão da língua, queixo proeminente, lábio superior fino, dentes espaçados ·8 Redução da pigmentação cutânea, com pele mais clara do que o padrão familiar e maior freqüência de cabelos finos e loiros e olhos claros ·9 Estrabismo (40% dos casos) e, mais raramente (10%), desvio na coluna (escoliose) ·10 Crises epilépticas, especialmente ausências, associadas a padrão característico do eletroencefalograma (EEG)
Aconselhamento Genético
Os exames genéticos são particularmente importantes e vários testes laboratoriais são necessários para o diagnóstico e aconselhamento genético. Os exames são:
Análise cariótipica, através de técnicas tradicionais e de citogenética molecular;
Testes específicos de DNA. Em cerca de 80% dos pacientes, o teste de DNA chamado padrão de metilação confirma a suspeita diagnóstica. Em 20% dos pacientes são necessários novos testes de DNA na procura de alterações genéticas que possam estar presentes no gene (ou genes) causador da Síndrome de Angelman.
Os mecanismos genéticos principais que causam a SA são deleção (perda) de material genético do cromossomo 15 ou dissomia uniparental (herança de dois cromossomos 15 de um mesmo progenitor). Nesses casos, o risco dos pais virem a ter outra criança afetada é muito baixo, 1%. Nos demais casos o risco do casal vir a ter outras crianças com a síndrome pode ser maior, chegando a 50% (risco alto) e deve ser calculado para a família.
Fonte: ASA – Associação da Síndrome de Angelman, organização fundada em 6 de agosto de 1997, por um grupo de pais de crianças portadoras de SA e simpatizantes da causa, ao constatarem o muito que havia por fazer. Maiores informações sobre a entidade, podem ser encontradas na HP da ASA, na pasta Organizações Associadas, deste site.
O que pode ser feito uma vez diagnosticada a SA?
A importância do diagnóstico da Síndrome de Angelman está em:
1 Poder-se antecipar a ocorrência de problemas característicos desta doença, tais como: distúrbios do sono e crises convulsivas, e, na medida do possível, tratá-los;·2 Poder orientar melhor as medidas de reabilitação, com fisioterapia, fonoterapia, terapia ocupacional e outros procedimentos;·3 Permitir orientar os pais quanto ao risco de repetição da SA na família, que deve ser avaliado em cada caso. Na maior parte das famílias, o risco de repetição de SA é muito pequeno, mas somente o estudo genético permite definir qual é ele;·4 Enfim, o diagnóstico permite a busca do porquê de um problema e facilita o planejamento do futuro do paciente.
Que exames podem ajudar no diagnóstico da SA?
Além do quadro clínico, alguns exames podem contribuir para o diagnóstico de SA. O EEG, avaliado por neurofisiologista com experiência nesta doença, poderá mostrar alterações fortemente sugestivas da SA. Os exames de imagem do crânio, como a tomografia computadorizada e a ressonância magnética, são normais ou apresentam alterações pouco específicas, pouco contribuindo para o diagnóstico de SA.
Os exames genéticos são particularmente importantes, sendo que em aproximadamente 80% dos casos, usando-se diferentes técnicas laboratoriais, pode-se confirmar a existência de deficiência em um dos cromossomos 15.
Em 20% dos casos, o diagnóstico é baseado apenas em dados clínicos e de outros exames complementares, sendo o estudo genético normal.
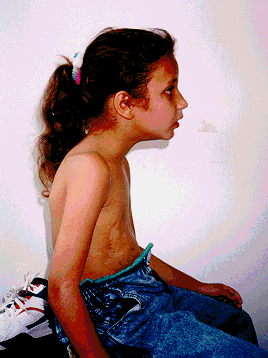
SÍNDROME DE ALGEMAN: Um Caso Raro e de Difícil TratamentoA Síndrome de Angelman, ocorre a cada 15.000 nascimentos e conta com poucos recursos de tratamento no Brasil.
A Síndrome de Angelman foi reconhecida em 1965 pelo médico inglês Dr. Harry Angelman, como um dano no cromossomo 15 herdado da mãe. Trata-se, portanto, de uma doença genética, sendo que em uma pequena parcela dos casos pode acontecer mais de uma vez na mesma família. O Dr. Angelman identificou, na época, três crianças com características comuns, tais como rigidez, dificuldades para andar, ausência de fala, riso excessivo e crises convulsivas.
A incidência exata é desconhecida e no Brasil pouco se conhece deste tema, embora recentemente já exista um site na Internet da Associação de Pais de portadores da síndrome Angelman, a ASA (http://asa.netway.com.br/). O site possui dois depoimentos de mães de crianças portadoras, com a descrição das dificuldades que sofreram para diagnosticar e tratar seus filhos. Além disso, a página conta com endereços úteis e uma série de outras informações a respeito da Síndrome. No Hospital de Clínicas de São Paulo, no ambulatório de genética, existem atualmente 10 casos catalogados, e outros 15 ou 20 em fase de diagnóstico.
Nos Estados Unidos e no Canadá também existe uma Fundação, a Angelman Syndrome Foundation. Nesta fundação americana existem cerca de 1000 indivíduos portadores catalogados. Sabe-se também que ao redor de todo o mundo existem casos diagnosticados da Síndrome de Angelman em diversas raças. Estima-se que ocorra um caso a cada 15 ou 30 mil nascimentos.
Normalmente, a Síndrome não é facilmente diagnosticada pelo pediatra clínico, em função da sua baixa incidência. Suspeitando de algum transtorno genético, os pacientes costumam ser encaminhados para os setores de neurologia e genética dos grandes hospitais, como Clínicas ou a Escola Paulista de Medicina, em São Paulo. “O diagnóstico é então confirmado por testes de DNA e estudo do cariótipo. Quem faz este diagnóstico é o Serviço de Aconselhamento Genético do Departamento de Biologia do Instituto de Biociências da Universidade de São Paulo” explica a Professora e Dra. Célia P. Koiffmann, do Instituto de Biociências. O Laboratório Fleury também encaminha o sangue para diagnóstico, que é feito então nos Estados Unidos.
A Síndrome é bastante difícil de ser reconhecida no recém-nascido ou na infância, uma vez que os problemas de desenvolvimento são inespecíficos neste período. Conhecendo-se as características da Síndrome, no entanto, é possível, mesmo para os pais, reconhecer uma criança portadora entre três e sete anos, quando os sintomas se fazem mais evidentes, embora já seja possível observar algum atraso no desenvolvimento a partir de 6 ou 12 meses de idade. De maneira geral, a gravidez de uma criança portadora da Síndrome de Angelman é normal.
Entre os sintomas clínicos consistentes, isto é, que estão presentes em 100% dos casos, levantados no documento “Síndrome de Angelman: Consenso por um critério diagnóstico”, de American Journal of Medical Genetics, publicado em 1995, constam, entre outros: o atraso do desenvolvimento, funcionalmente severo; a incapacidade de falar, com nenhum ou quase nenhum uso de palavras, identificando-se uma maior capacidade de compreensão do que de expressão verbal. São observados problemas de movimento e equilíbrio, com incapacidade de coordenação dos movimentos musculares voluntários ao andar e/ou movimento trêmulo dos membros. É freqüente qualquer combinação de riso e sorriso, com uma aparência feliz (embora este sorriso permanente seja apenas uma expressão motora, e não uma forma de comunicação, explica o Dr. Paulo Plaggerp, pediatra do Ambulatório de Genética do Hospital de Clínicas), associada a uma personalidade facilmente excitável, com movimentos aleatórios das mãos, hipermotrocidade e incapacidade de manter a atenção.
Em mais de 80% dos casos é observado o atraso no crescimento do perímetro cefálico, normalmente produzindo microcefalia (absoluta ou relativa) em torno dos dois anos de idade. Crises convulsivas antes dos três anos de idade também são comuns. Nesta proporção, de 80% de incidência, observa-se eletroencefalograma anormal, com ondas de grande amplitude e picos lentos.
Em menos de 80% dos casos verificam-se outros sintomas associados, tais como estrabismo, hipopigmentação da pele e dos olhos, hipersensibilidade ao calor, mandíbula e língua proeminentes, problemas para succionar, baba freqüente, língua para fora da boca, boca grande, dentes espaçados, atração pela água, comportamento excessivo de mastigação, hiperatividade dos movimentos reflexos nos tendões, problemas para dormir e com a alimentação durante a infância, braços levantados e flexionados ao caminhar, define a Professora e Dra. Célia.
De acordo com o Dr. Paulo Plaggerp, do Ambulatório de Genética do Hospital de Clínicas de São Paulo, não há tratamento disponível para os portadores da Síndrome de Angelman. É possível apenas dar tratamento de suporte, ou psicossomático, procurando amenizar os sintomas e melhorar a qualidade de vida dos portadores.
Convulsões
Mais de 90% dos casos reportaram ter convulsões, mas esta pode ser uma superestimativa porque os informes médicos tendem a estudar os casos mais severos. As convulsões podem ser de qualquer tipo e requererem medicamentos anticonvulsivos.
Caminhada e problemas de movimentos
Movimentos do tronco e membros são descritos nos primeiros anos e movimentos nervosos ou tremores podem estar presentes nos primeiros seis meses de vida. A conseqüência é que etapas normais da motricidade grossa ficam atrasadas; normalmente se sentam depois dos doze meses e não andam até os três ou quatro anos.
As crianças mais seriamente afetadas podem ser rígidas, como um robô, e inseguras para caminhar; aproximadamente 10% não chegam a andar.
Hiperatividade
A hiperatividade é provavelmente a conduta mais típica dos portadores da Síndrome de Angelman. Todas as crianças portadoras apresentam algum componente, afetando igualmente ambos os sexos. Em casos extremos, o movimento constante pode causar acidentes ou contusões. Condutas como agarrar, beliscar e morder crianças maiores também ocorrem pela atividade hipermotora. Terapias persistentes e consistentes de mudança de conduta ajudam a diminuir ou eliminar as condutas socialmente não desejadas.
Riso e felicidade
Não se sabe por que o riso é tão freqüente nos portadores da Síndrome. Estudos do cérebro não tem mostrado nenhum defeito que leve a pensar num campo anormal para o riso. O riso parece ser, fundamentalmente, uma manifestação de expressão motora, e não uma expressão de sentimentos ou uma forma de comunicação, como já explicado. A maioria de reações aos estímulos físicos e mentais se acompanha de riso. Ainda que as crianças portadoras experimentem uma variedade de emoções, aparentemente predomina a felicidade.
Fala e linguagem
uns portadores parecem ter bastante compreensão, levando a crer que seriam capazes de falar. Porém, mesmo nos de mais alto nível de desenvolvimento, a linguagem não se desenvolve. Em geral, relata-se que alguns indivíduos chegam a falar de uma a três palavras. Em um estudo de 47 indivíduos, computou-se que 39% falavam até quatro palavras. Os que apresentam capacidades verbais mais altas, chegam a usar de 10-20 palavras. A capacidade de linguagem não verbal varia muito, sendo que os mais desenvolvidos são capazes de aprender alguns sinais e usá-los como ajuda de expressão, juntamente com outros meios de comunicação baseados em imagens, como murais e lousas.
Atraso mental e desenvolvimento
A comprovação de que o desenvolvimento está comprometido verifica-se pela falta de atenção, hiperatividade, falta de linguagem verbal e controle motor. Sabe-se que as capacidades cognitivas nos portadores da Síndrome são mais altas que as indicadas pelos testes de desenvolvimento. O que torna isto evidente é a área que diferencia a linguagem compreensiva da linguagem falada. Diante disso, as crianças portadoras da Síndrome diferenciam-se de outros quadros de atraso mental severo e os jovens adultos são, normalmente, socialmente adaptados e respondem à maioria dos sinais pessoais e interações. Eles participam de atividades de grupo, afazeres domésticos e nas atividades e responsabilidades do convívio diário, embora nunca se tornem independentes. Como os outros, eles desfrutam da maioria das atividades recreativas como televisão, esportes, etc. É preciso ressaltar, no entanto, que este nível de interação também depende do nível de dano causado pela Síndrome.
Hipopigmentação
Por vezes, ocorre hipopigmentação da pele e dos olhos. Em alguns casos, tamanha a força desse fenômeno, pode-se confundi-lo com alguma manifestação de albinismo; há hipersensibilidade aos raios solares, o que requer o uso de protetores potentes.
Estrabismo e albinismo ocular
Estudos de pacientes com Síndrome de Angelman demonstram que a incidência de estrabismo se dá em 30-60% dos casos. Este problema parece ser mais comum em crianças com hipopigmentação ocular, dado que o pigmento na retina é crucial para o desenvolvimento normal das ramificações do nervo óptico.
Transtornos do sono
A diminuição da necessidade de dormir e ciclos anormais de sono/vigília é característica de portadores da Síndrome. O uso de sedativos pode ser útil, caso a vigília esteja interferindo demais na vida dos membros da família.
Adolescência
Durante a adolescência, a puberdade pode estar atrasada de um a três anos, mas a maturação sexual ocorre normalmente. A saúde física parece ser notavelmente boa. A expectativa de vida não parece ser muito menor, tem-se notícia de pessoas na faixa dos 60 anos e muitas com mais de quatro décadas de vida.
Encaminhamento
Sem dúvida, com uma intervenção intensa nos seus perfis de conduta e estimulação, a criança portadora começa a superar os problemas mais graves e os progressos no desenvolvimento passam a ser visíveis.
As crianças com pouca estabilidade de andar podem obter benefícios em fisioterapia. A terapia ocupacional pode ajudar a melhorar a motricidade fina e controlar a conduta motoro-bucal. Terapias de comunicação e fonoaudiologia são essenciais. Técnicas de modificação de conduta, tanto em casa, quanto no colégio, podem permitir que a criança possa desenvolver, ela mesma, a capacidade de realizar a maioria das tarefas relacionadas com o comer, o vestir e atividades de casa.
- Autor: Maria Lúcia Coelho Dias
Sou professora e minha filha tem SA e fico muito perdida como proceder com ela em atividades pedagógicas
Na escola onde trabalho temos uma aluna com síndrome de angelmen, e gostaríamos de receber informações de como tr
abalhar e melhor estimula-la.
Se inscreva na nossa newsletter!